
Tramadol, sold under the brand name Ultram among others, is an opioid pain medication used to treat moderate to moderately severe pain. When taken by mouth in an immediate-release formulation, the onset of pain relief usually begins within an hour. It is also available by injection. It may be sold in combination with paracetamol (acetaminophen) or as longer-acting formulations.
As is typical of opioids, common side effects include constipation, itchiness, and nausea. Serious side effects may include seizures, increased risk of serotonin syndrome, decreased alertness, and drug addiction. A change in dosage may be recommended in those with kidney or liver problems. It is not recommended in those who are at risk of suicide or in those who are pregnant. While not recommended in women who are breastfeeding, those who take a single dose should not generally stop breastfeeding. Tramadol is converted in the liver to O-desmethyltramadol (desmetramadol), an opioid with stronger binding to the μ-opioid receptor. Tramadol is also a serotonin–norepinephrine reuptake inhibitor (SNRI).
Tramadol was patented in 1963 and launched under the name “Tramal” in 1977 by the West German pharmaceutical company Grünenthal GmbH. In the mid-1990s, it was approved in the United Kingdom and the United States. It is available as a generic medication and marketed under many brand names worldwide. In 2018, it was the 25th most commonly prescribed medication in the United States, with more than 24 million prescriptions.
Medical uses

Generic tramadol HCl tablets marketed by Amneal Pharmaceuticals

Tramadol HCl for injection
Tramadol (a schedule IV drug in the US) is used primarily to treat mild to severe pain, both acute and chronic. There is moderate evidence for use as a second-line treatment for fibromyalgia but is not FDA approved for this use, however, its use is approved for treatment of fibromyalgia as a secondary painkiller by the NHS.
Its analgesic effects take about one hour to come into effect and 2 to 4 h to peak after oral administration with an immediate-release formulation. On a dose-by-dose basis, tramadol has about one-tenth the potency of morphine (thus 100 mg is commensurate with 10 mg morphine but may vary) and is practically equally potent when compared with pethidine and codeine . For pain moderate in severity, its effectiveness is equivalent to that of codeine at low doses, and hydrocodone at very high doses; for severe pain it is less effective than morphine.
These painkilling effects last about 6 h. The potency of analgesia varies considerably as it depends on an individual’s genetics. People with specific variants of CYP2D6 enzymes may not produce adequate amounts of the active metabolite (desmetramadol) for effective pain control.
Sleep medicine physicians sometimes prescribe tramadol (or other opiate medications) for refractory restless legs syndrome (RLS); that is, RLS that does not respond adequately to treatment with first-line medications such as dopamine agonists (like pramipexole) or alpha-2-delta (α2δ) ligands (gabapentinoids), often due to augmentation.
Contraindications
Tramadol may not provide adequate pain control for individuals with certain genetic variants of CYP2D6 enzymes as they metabolize tramadol to the inactive molecule. These genetic polymorphisms are not currently routinely tested for in clinical practice.
Pregnancy and lactation
Tramadol’s use in pregnancy is generally avoided, as it may cause some reversible withdrawal effects in the newborn. A small prospective study in France found, while an increased risk of miscarriages existed, no major malformations were reported in the newborn. Its use during lactation is also generally advised against, but a small trial found that infants breastfed by mothers taking tramadol were exposed to about 2.88% of the dose the mothers were taking. No evidence of this dose having a harmful effect on the newborn was seen.
Labor and delivery
Its use as an analgesic during labor is not advised due to its long onset of action (1 hour). The ratio of the mean concentration of the drug in the fetus compared to that of the mother when it is given intramuscularly for labor pains has been estimated to be 1:94.
Children
Its use in children is generally advised against, although it may be done under the supervision of a specialist. On 21 September 2015, the FDA started investigating the safety of tramadol in use in persons under the age of 17. The investigation was initiated because some of these people have experienced slowed or difficult breathing. The FDA lists age under 12 years old as a contraindication.
Elderly
The risk of opioid-related adverse effects such as respiratory depression, falls, cognitive impairment and sedation is increased. Tramadol may interact with other medications and increase the risk for adverse events.
Liver and kidney failure
The drug should be used with caution in those with liver or kidney failure, due to metabolism in the liver (to the active molecule desmetramadol) and elimination by the kidneys.
Side effects
The most common adverse effects of tramadol include nausea, dizziness, dry mouth, indigestion, abdominal pain, vertigo, vomiting, constipation, drowsiness, and headache. Other side effects may result from interactions with other medications. Tramadol has the same dose-dependent adverse effects as morphine including respiratory depression.
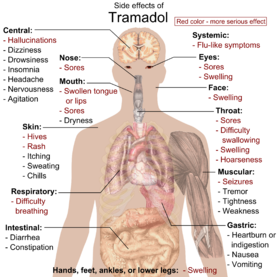
Main side effects of tramadol: Red color denotes more serious effects, requiring immediate contact with health provider.
Dependence and withdrawal
Long-term use of high doses of tramadol causes physical dependence and withdrawal syndrome. These include both symptoms typical of opioid withdrawal and those associated with serotonin–norepinephrine reuptake inhibitor (SNRI) withdrawal; symptoms include numbness, tingling, paresthesia, and tinnitus. Psychiatric symptoms may include hallucinations, paranoia, extreme anxiety, panic attacks, and confusion. In most cases, tramadol withdrawal will set in 12–20 hours after the last dose, but this can vary. Tramadol withdrawal typically lasts longer than that of other opioids. Seven days or more of acute withdrawal symptoms can occur as opposed to typically 3 or 4 days for other codeine analogues.
Overdose
Recognised risk factors for tramadol overdose include depression, addiction, and seizures. Naloxone only partially reverses the toxic effects of tramadol overdose and may increase the risk of seizures.
Deaths with tramadol overdose have been reported and are increasing in frequency in Northern Ireland; the majority of these overdoses involves other drugs including alcohol. There were 254 tramadol-related deaths in England and Wales in 2013, and 379 in Florida in 2011. In 2011, 21,649 emergency room visits in the United States were related to tramadol.
Interactions
Tramadol can interact with other medications with similar mechanisms of action.
Tramadol acts as a serotonin-norephinephrine reuptake inhibitor and thus can interact with other serotonergic medications (selective serotonin reuptake inhibitors, serotonin–norepinephrine reuptake inhibitors, tricyclic antidepressants, triptans, cough and cold medications containing dextromethorphan, herbal products containing St. John’s wort, and medications that inhibit the metabolism of serotonin, such as monoamine oxidase inhibitors) and, in combination, may lead to serotonin syndrome. It may also make some serotonergic antagonist anti-emetic medications (ondansetron) less effective.
Tramadol also acts as an opioid agonist and thus can increase the risk for side effects when used with other opioid analgesics (such as morphine, pethidine, tapentadol, oxycodone, and fentanyl).
Tramadol is metabolized by CYP2D6 enzymes which contribute to the metabolism of approximately 25% of all medications. Any medications with the ability to inhibit or induce these enzymes may interact with tramadol.
Tramadol increases the risk for seizures by lowering the seizure threshold. Using other medications that lower seizure threshold (such as antipsychotic medications or amphetamines), further increases this risk.
Pharmacology
Mechanism of action
Tramadol induces analgesic effects through a variety of different targets on the noradrenergic system, serotoninergic system and opioid receptors system. Tramadol exists as a racemic mixture, the positive enantiomer inhibits serotonin reuptake while the negative enantiomer inhibits noradrenaline re-uptake, by binding to and blocking the transporters. Tramadol has also been shown to act as a serotonin releasing agent. Both enantiomers of tramadol are agonists of the μ-opioid receptor and its M1 metabolite, O-desmetramadol, is also a μ-opioid receptor agonist but is 6 times more potent than tramadol itself. All these effects work synergistically to induce analgesia.
| Site | Tramadol | DSMT | Species | Ref |
|---|---|---|---|---|
| MOR | 1,600–12,486 2,120–8,300 ≥1,000 (EC50) |
5.4–18.6 17 ((+)) ≥240 (EC50) |
Human Rat Human |
|
| DOR | >10,000 57,600–100,000 |
≥2,900 690 (+)) |
Human Rat |
|
| KOR | >10,000 42,700–81,000 |
≥450 1,800 (+)) |
Human Rat |
|
| SERT | ~900 (IC50) 992–1,190 |
>20,000 (IC50) 2,980 ((−)) (IC50) |
Human Rat |
|
| NET | 14,600 785 |
1,080 (−) (IC50) >860 (IC50) |
Human Rat |
|
| DAT | >100,000 | >20,000 | Rat | |
| 5-HT1A | >20,000 | >20,000 | Rat | |
| 5-HT2A | >20,000 | >20,000 | Rat | |
| 5-HT2C | 1,000 (IC50) | 1,300 (IC50) | Rat | |
| 5-HT3 | >20,000 | >20,000 | Rat | |
| NK1 | IA | ? | Rat | |
| M1 | >20,000 3,400 (IC50) |
>20,000 2,000 (IC50) |
Rat Multiple |
|
| M2 | ND | ND | ND | ND |
| M3 | 1,000 (IC50) | IA | Human | |
| M4 | ND | ND | ND | ND |
| M5 | ND | ND | ND | ND |
| α7 | 7,400 | ND | Chicken | |
| σ1 | >10,000 | ND | Rat | |
| σ2 | >10,000 | ND | Rat | |
| NMDAR | 16,400 (IC50) | 16,500 (IC50) | Human | |
| NMDAR (MK-801) |
>20,000 | >20,000 | Rat | |
| GABAA | >100,000 (IC50) | >100,000 (IC50) | Human | |
| GlyR | >100,000 (IC50) | >100,000 (IC50) | Human | |
| TRPA1 | 100– 10,000 (SI) |
1,000– 10,000 (SI) |
Human | |
| TRPV1 | >10,000 (IC50) | >10,000 (IC50) | Human | |
| Values are Ki (nM), unless otherwise noted. The smaller the value, the more strongly the drug binds to the site. | ||||
| Action | Value |
|---|---|
| 5-HT reuptake | 1,820 |
| 5-HT release | >10,000 |
| NE reuptake | 2,770 |
| NE release | >10,000 |
| DA reuptake | >10,000 |
| DA release | >10,000 |
| Values for reuptake inhibition are Ki (nM) and for release induction are EC50 (nM). | |
Tramadol has been found to possess these actions:
- Agonist of the μ-opioid receptor (MOR) and to a far lesser extent of the δ-opioid receptor (DOR) and κ-opioid receptor (KOR)
- Serotonin reuptake inhibitor (SRI) and norepinephrine reuptake inhibitor; hence, an SNRI
- Serotonin 5-HT2C receptor antagonist
- M1 and M3 muscarinic acetylcholine receptor antagonist
- α7 nicotinic acetylcholine receptor antagonist
- NMDA receptor antagonist (very weak)
- TRPA1 inhibitor
Tramadol acts on the opioid receptors through its major active metabolite desmetramadol, which has as much as 700-fold higher affinity for the MOR relative to tramadol. Moreover, tramadol itself has been found to possess no efficacy in activating the MOR in functional activity assays, whereas desmetramadol activates the receptor with high intrinsic activity (Emax equal to that of morphine). As such, desmetramadol is exclusively responsible for the opioid effects of tramadol. Both tramadol and desmetramadol have pronounced selectivity for the MOR over the DOR and KOR in terms of binding affinity.
Tramadol is well-established as an SRI. In addition, a few studies have found that it also acts as a serotonin releasing agent (1–10 μM), similar in effect to fenfluramine. The serotonin releasing effects of tramadol could be blocked by sufficiently high concentrations of the serotonin reuptake inhibitor 6-nitroquipazine, which is in accordance with other serotonin releasing agents such as fenfluramine and MDMA. However, two more recent studies failed to find a releasing effect of tramadol at respective concentrations up to 10 and 30 μM. In addition to serotonergic activity, tramadol is also a norepinephrine reuptake inhibitor. It is not a norepinephrine releasing agent. Tramadol does not inhibit the reuptake or induce the release of dopamine.
A positron emission tomography imaging study found that single oral 50-mg and 100-mg doses of tramadol to human volunteers resulted in 34.7% and 50.2% respective mean occupation of the serotonin transporter (SERT) in the thalamus. The estimated median effective dose (ED50) for SERT occupancy hence was 98.1 mg, which was associated with a plasma tramadol level of about 330 ng/ml (1,300 nM). The estimated maximum daily dosage of tramadol of 400 mg (100 mg q.i.d.) would result in as much as 78.7% occupancy of the SERT (in association with a plasma concentration of 1,220 ng/ml or 4,632 nM). This is close to that of SSRIs, which occupy the SERT by 80% or more.
Peak plasma concentrations during treatment with clinical dosages of tramadol have generally been found to be in the range of 70 to 592 ng/ml (266–2,250 nM) for tramadol and 55 to 143 ng/ml (221–573 nM) for desmetramadol. The highest levels of tramadol were observed with the maximum oral daily dosage of 400 mg per day divided into one 100-mg dose every 6 hours (i.e., four 100-mg doses evenly spaced out per day). Some accumulation of tramadol occurs with chronic administration; peak plasma levels with the maximum oral daily dosage (100 mg q.i.d.) are about 16% higher and the area-under-the-curve levels 36% higher than following a single oral 100-mg dose. Positron emission tomography imaging studies have reportedly found that tramadol levels are at least four-fold higher in the brain than in plasma. Conversely, brain levels of desmetramadol “only slowly approach those in plasma”. The plasma protein binding of tramadol is only 4 to 20%; hence, almost all tramadol in circulation is free, thus bioactive.
Correspondence to effects
Co-administration of quinidine, a potent CYP2D6 enzyme inhibitor, with tramadol, a combination which results in markedly reduced levels of desmetramadol, was found not to significantly affect the analgesic effects of tramadol in human volunteers. However, other studies have found that the analgesic effects of tramadol are significantly decreased or even absent in CYP2D6 poor metabolizers. The analgesic effects of tramadol are only partially reversed by naloxone in human volunteers, hence indicating that its opioid action is unlikely the sole factor; tramadol’s analgesic effects are also partially reversed by α2-adrenergic receptor antagonists such as yohimbine, the 5-HT3 receptor antagonist ondansetron, and the 5-HT7 receptor antagonists SB-269970 and SB-258719. Pharmacologically, tramadol is similar to tapentadol and methadone in that it not only binds to the MOR, but also inhibits the reuptake of serotonin and norepinephrine due to its action on the noradrenergic and serotonergic systems, such as its “atypical” opioid activity.
Tramadol has inhibitory actions on the 5-HT2C receptor. Antagonism of 5-HT2C could be partially responsible for tramadol’s reducing effect on depressive and obsessive–compulsive symptoms in patients with pain and co-morbid neurological illnesses. 5-HT2C blockade may also account for its lowering of the seizure threshold, as 5-HT2C knockout mice display significantly increased vulnerability to epileptic seizures, sometimes resulting in spontaneous death. However, the reduction of seizure threshold could be attributed to tramadol’s putative inhibition of GABAA receptors at high doses (significant inhibition at 100 μM). In addition, desmetramadol is a high-affinity ligand of the DOR, and activation of this receptor could be involved in tramadol’s ability to provoke seizures in some individuals, as DOR agonists are well known for inducing seizures.
Nausea and vomiting caused by tramadol are thought to be due to activation of the 5-HT3 receptor via increased serotonin levels. In accordance, the 5-HT3 receptor antagonist metoclopramide can be used to treat tramadol-associated nausea and vomiting. Tramadol and desmetramadol themselves do not bind to the 5-HT3 receptor.
Pharmacokinetics

Desmetramadol.
Tramadol undergoes hepatic metabolism via the cytochrome P450 isozyme CYP2B6, CYP2D6, and CYP3A4, being O– and N-demethylated to five different metabolites. Of these, desmetramadol (O-desmethyltramadol) is the most significant, since it has 200 times the μ-affinity of (+)-tramadol, and furthermore has an elimination half-life of 9 hours, compared with 6 hours for tramadol itself. As with codeine, in the 6% of the population who have reduced CYP2D6 activity (hence reducing metabolism), a reduced analgesic effect is seen. Those with decreased CYP2D6 activity require a dose increase of 30% to achieve the same degree of pain relief as those with a normal level of CYP2D6 activity.
Phase II hepatic metabolism renders the metabolites water-soluble, which are excreted by the kidneys. Thus, reduced doses may be used in renal and hepatic impairment.
Its volume of distribution is around 306 l after oral administration and 203 l after parenteral administration.
Chemistry
Tramadol is marketed as a racemic mixture of both R– and S-stereoisomers, because the two isomers complement each other’s analgesic activities. The (+)-isomer is predominantly active as an opiate with a higher affinity for the µ-opiate receptor (20 times higher affinity than the (-)-isomer).
Synthesis and stereoisomerism
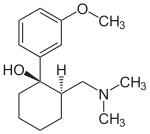 |
 |
| (1R,2R)-tramadol | (1S,2S)-tramadol |
 |
 |
| (1R,2S)-tramadol | (1S,2R)-tramadol |
The chemical synthesis of tramadol is described in the literature. Tramadol has two stereogenic centers at the cyclohexane ring. Thus, 2-(dimethylaminomethyl)-1-(3-methoxyphenyl)cyclohexanol may exist in four different configurational forms:
- (1R,2R)-isomer
- (1S,2S)-isomer
- (1R,2S)-isomer
- (1S,2R)-isomer
The synthetic pathway leads to the racemate (1:1 mixture) of (1R,2R)-isomer and the (1S,2S)-isomer as the main products. Minor amounts of the racemic mixture of the (1R,2S)-isomer and the (1S,2R)-isomer are formed as well. The isolation of the (1R,2R)-isomer and the (1S,2S)-isomer from the diastereomeric minor racemate is realized by the recrystallization of the hydrochlorides. The drug tramadol is a racemate of the hydrochlorides of the (1R,2R)-(+)- and the (1S,2S)-(−)-enantiomers. The resolution of the racemate was described employing (R)-(−)- or (S)-(+)-mandelic acid. This process does not find industrial application, since tramadol is used as a racemate, despite known different physiological effects of the (1R,2R)- and (1S,2S)-isomers, because the racemate showed higher analgesic activity than either enantiomer in animals and in humans.
Detection in biological fluids
Tramadol and desmetramadol may be quantified in blood, plasma or serum to monitor for abuse, confirm a diagnosis of poisoning or assist in the forensic investigation of a sudden death. Most commercial opiate immunoassay screening tests do not cross-react significantly with tramadol or its major metabolites, so chromatographic techniques must be used to detect and quantitate these substances. The concentration of desmetramadol in the blood or plasma of a person who has taken tramadol is generally 10–20% those of the parent drug.
Society and culture
Formulations
Available dosage forms include liquids, syrups, drops, elixirs, effervescent tablets and powders for mixing with water, capsules, tablets including extended-release formulations, suppositories, compounding powder, and injections.
Patent history
The U.S. Food and Drug Administration (FDA) approved tramadol in March 1995, and an extended-release (ER) formulation in September 2005. ER Tramadol was protected by US patents nos. 6,254,887 and 7,074,430. The FDA listed the patents’ expiration as 10 May 2014. However, in August 2009, US District Court for the District of Delaware ruled the patents invalid, a decision upheld the following year by the Court of Appeals for the Federal Circuit. Manufacture and distribution of generic equivalents of Ultram ER in the United States was therefore permitted prior to the expiration of the patents.
Legal status
Effective 18 August 2014, tramadol has been placed into Schedule IV of the federal Controlled Substances Act in the United States. Before that, some US states had already classified tramadol as a Schedule IV controlled substance under their respective state laws.
Tramadol is classified in Schedule 4 (prescription only) in Australia, rather than as a Schedule 8 Controlled Drug (Possession without authority illegal) like most other opioids.
Effective May 2008, Sweden classified tramadol as a controlled substance in the same category as codeine and dextropropoxyphene, but allows a normal prescription to be used.
The UK classified tramadol as a Class C, Schedule 3 controlled drug on 10 June 2014, but exempted it from the safe custody requirement.
Misuse
Illicit use of the drug is thought to be a major factor in the success of the Boko Haram terrorist organization. When used at higher doses, the drug “can produce similar effects to heroin.” Said one former member, “whenever we took tramadol, nothing mattered to us anymore except what we were sent to do because it made us very high and very bold, it was impossible to go on a mission without taking it.” Tramadol is also used as a coping mechanism in the Gaza Strip.
Research
Investigational uses
- Diabetic neuropathy
- Antidepressant
- Postherpetic neuralgia
- Premature ejaculation
- Adjunct to local anaesthesia
False findings about sources in nature
In 2013, researchers reported that tramadol was found in relatively high concentrations (1%+) in the roots of the African pin cushion tree (Nauclea latifolia). In 2014, however, it was reported that the presence of tramadol in the tree roots was the result of tramadol having been administered to cattle by farmers in the region: tramadol and its metabolites were present in the animals’ excreta, which contaminated the soil around the trees. Therefore, tramadol and its mammalian metabolites were found in tree roots in the far north of Cameroon, but not in the south where it is not administered to farm animals.
A 2014 editorial in Lab Times online contested the notion that tramadol in tree roots was the result of anthropogenic contamination, stating that samples were taken from trees which grew in national parks, where livestock were forbidden; it also quoted researcher Michel de Waard, who stated that “thousands and thousands of tramadol-treated cattle sitting around a single tree and urinating there” would be required to produce the concentrations discovered.
In 2015, radiocarbon analysis confirmed that the tramadol found in N.latifolia roots could not be plant-derived and was of synthetic origin.
Veterinary medicine
Tramadol may be used to treat post-operative, injury-related, and chronic (e.g., cancer-related) pain in dogs and cats as well as rabbits, coatis, many small mammals including rats and flying squirrels, guinea pigs, ferrets, and raccoons.
| Species | Half-life (h) for parent drug | Half-life (h) for desmetramadol | Maximum plasma concentration (ng/mL) for parent drug | Maximum plasma concentration (ng/mL) for desmetramadol |
|---|---|---|---|---|
| Camel | 3.2 (IM), 1.3 (IV) | – | 0.44 (IV) | – |
| Cat | 3.40 (oral), 2.23 (IV) | 4.82 (oral), 4.35 (IV) | 914 (oral), 1323 (IV) | 655 (oral), 366 (IV) |
| Dog | 1.71 (oral), 1.80 (IV), 2.24 (rectal) | 2.18 (oral), 90-5000 (IV) | 1402.75 (oral) | 449.13 (oral), 90–350 (IV) |
| Donkey | 4.2 (oral), 1.5 (IV) | – | 2817 (oral) | – |
| Goat | 2.67 (oral), 0.94 (IV) | – | 542.9 (oral) | – |
| Horses | 1.29–1.53 (IV), 10.1 (oral) | 4 (oral) | 637 (IV), 256 (oral) | 47 (oral) |
| Llama | 2.54 (IM), 2.12 (IV) | 7.73 (IM), 10.4 (IV) | 4036 (IV), 1360 (IM) | 158 (IV), 158 (IM) |